
Immunneuropathien sind eine wichtige Gruppe der behandelbaren peripheren Neuropathien, die durch eine Fehlregulation des Immunsystems verursacht werden. Dazu gehören die akute inflammatorische demyelinisierende Polyneuropathie (AIDP) oder Guillian-Barré-Syndrom (GBS), die chronisch-inflammatorische demyelinisierende Polyneuropathie (CIDP), paranodale Antikörper-vermittelte Neuropathien sowie weitere Formen wie die Anti-MAG-Neuropathie und die multifokale motorische Neuropathie (MMN).
CIDP und ihre Varianten
Die CIDP ist die häufigste chronische Immunneuropathie und zeigt typischerweise einen schleichenden oder auch schubförmigen Verlauf mit motorischen und sensiblen Ausfällen. Neben der klassischen Form gibt es Varianten wie die CIDP mit monoklonaler Gammopathie (CIDP-MGUS), das Lewis-Sumner-Syndrom (MADSAM), die distale erworbene demyelinisierende symmetrische Neuropathie (DADS) und sensible ataktische CIDP.
Paranodale Neuropathien
werden durch Antikörper gegen Proteine im Bereich der Ranvier’schen Schnürringe (z. B. Caspr1, Contactin-1, Neurofascin-155) verursacht. Diese Formen sind oft schwerer therapierbar und erfordern gezielte Diagnostik sowie eine differenzierte Therapie. Die Identifizierung dieser Antikörper ist entscheidend für die Wahl der richtigen Therapie.
Weitere Antikörper-vermittelte Neuropathien
Neben den paranodalen Antikörpern gibt es weitere Formen wie die Anti-MAG-Neuropathie (Antikörper gegen Myelin-Associated Glycoprotein), die typischerweise mit einer IgM-Gammopathie assoziiert ist sowie vaskulitische Neuropathien und Neuropathien im Rahmen von chronisch-entzündlichen Darmerkrankungen, Sarkoidose oder rheumatischen Erkrankungen.
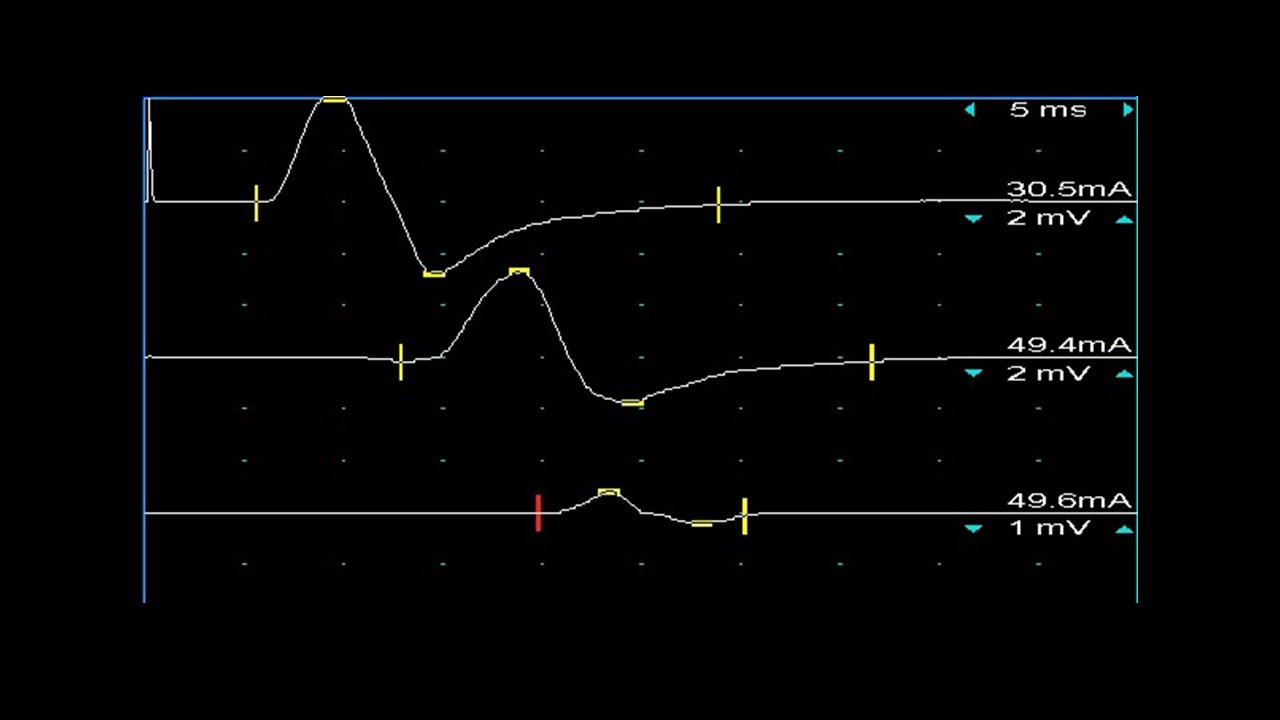
Das diagnostische Vorgehen beinhaltet neben der klinischen Untersuchung vor allem die elektrophysiologische Kategorisierung des Nervenschädigungsmuster. Im Vordergrund stehen dabei ganz überwiegend diffuse Schädigungen oder umschriebene Läsionen (sognannte Leitungsblöcke) der Markscheide (Nervenhülle). Die Nervensonografie liefert wertvolle Informationen, insbesondere zur Abgrenzung gegenüber anderen Neuropathien liefern. Neben Labor-und serologischen Untersuchungen (bestimmte Autoantikörper, Paraproteinämie) spielt die Untersuchung des Liquors (Nervenflüssigkeit) eine wichtige Rolle, das sich hier häufig eine Erhöhung des Liquoreiweiß findet, als Ausdruck einer “Entzündung” der nervenwurzelnahen Abschnitte der Nerven.
In Abhängigkeit von der genauen Diagnose kommen immunsuppressive oder immunmodulierende Therapien zum Einsatz. Das Spektrum reicht von Kortison über intravenöse oder subkutane Immunglobuline über etablierte Immunsuppressiva bis hin zu spezifischen monoklonalen Antikörpertherapien (z.B. FcRn-Inhibitoren)