
Das diagnostisch charakteristische Merkmal ist die pathologische Übererregbarkeit der Muskelzellmembran, die auf Störungen in unterschiedlichen Ionenkanälen zurückgeht, welche die elektrische Aktivität der Muskulatur regulieren. Ein bemerkenswertes klinisches Phänomen bei nicht-dystrophen Myotinien ist die Muskelvergrößerung (Hypertrophie), insbesondere an Waden-, Oberschenkel- und Gesäßmuskulatur, was die Betroffenen muskulös erscheinen lässt, während die Kraft paradoxerweise oft eingeschränkt ist. Bei den dystrophen Myotonien findet sich hingegen eine Muskelverschmächtigung mit Muskelschwäche. Eine charakteristische Besonderheit vieler Myotonien ist das „warming-up-Phänomen", die Myotonie nimmt bei wiederholten Muskelkontraktionen ab. Das Elektromyogramm zeigt typische myotone Entladungen mit Crescendo / Decrescendo Modulation.
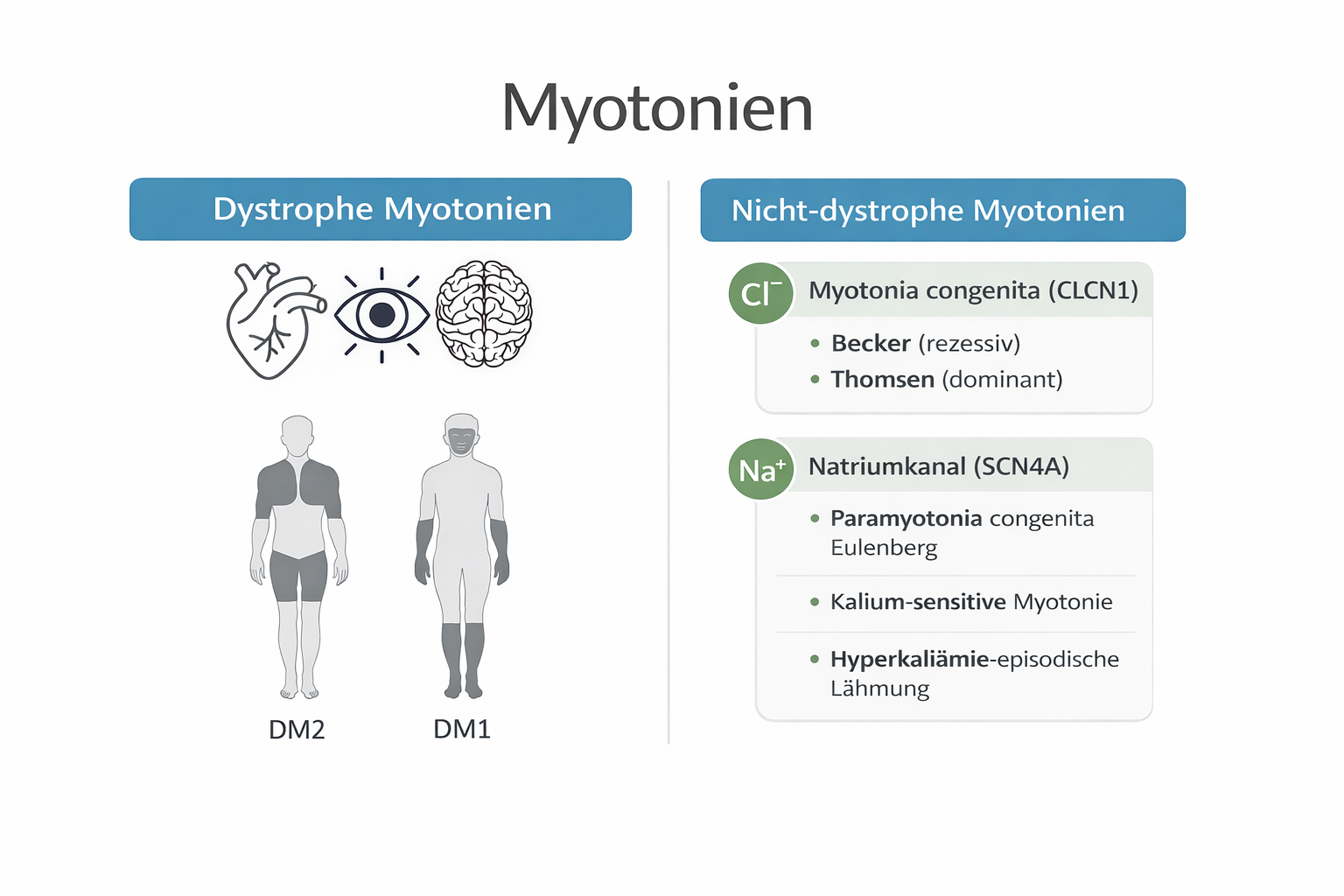
Dystrophe Myotonien oder multisystemische Myotonien durch weitere extramuskuläre Manifestationen:
- Myotone Dystrophie Typ 1 (DM1) – auch Curschmann-Steinert-Batten-Syndrom genannt, die häufigste Muskeldystrophie des Erwachsenenalters, charakterisiert durch progressive Paresen (Lähmungen) und Muskelschwund.
- Myotone Dystrophie Typ 2 (DM2) – auch als PROMM (Proximale Myotone Myopathie) bezeichnet, mit ähnlichen Merkmalen aber typischerweise später beginnend und einem anderen Verteilungsmuster der Lähmungen vorzugsweis im Gliedergürtel.
Nicht-dystrophe Myotonien, diese entstehen durch Defekte in Ionenkanälen und werden nach dem betroffenen Kanal-Typ eingeteilt:
Chloridkanal-Myotonien:
- Myotonia congenita Thomsen (dominant vererbte Form) – tritt bereits im frühesten Kindesalter in Erscheinung, mit milder bis mäßiger Muskelsteifigkeit und normalerweise ohne Muskelschwäche.
Myotonia congenita Becker (rezessiv vererbte Form) – beginnt typischerweise im Schulalter oder später, zeigt ausgeprägtere Myotonie und kann mit vorübergehender Muskelschwäche einhergehen.
Natriumkanal-Myotonien (Kalium-sensitive Myotonien), diese seltenen Formen zeigen eine charakteristische Verschlechterung durch orale Kaliumaufnahme oder Schwankungen im Kaliumspiegel
- Paramyotonia congenita Eulenburg – kälteabhängige Form, bei der Myotonie durch Kältexposition zunimmt
- Myotonia fluctuans – leichteste Form der Natriumkanal-Myotonien, mit intermittierender Steifigkeit, die erst mit zeitlicher Verzögerung nach Muskelarbeit auftritt
- Myotonia permanens – schwerste Form mit kontinuierlicher Muskelversteifung, die erhebliche funktionelle Beeinträchtigungen verursacht.
Die grundlegende Störung bei Myotonien besteht in einer Übererregbarkeit der Muskelzellmembran. Bei Chloridkanal-Myotonien führen Mutationen im Gen CLCN1 zu einer verminderten Chloridleitfähigkeit der Muskelfasermembran, die normalerweise etwa 80% der Gesamtleitfähigkeit ausmacht und für die Stabilisierung des Ruhemembranpotentials essentiell ist. Wenn die Chloridleitfähigkeit unter 30% fällt, entsteht die klinische Myotonie.
Bei Natriumkanal-Myotonien (Mutationen im SCN4A-Gen) liegt eine fehlende Inaktivierung der muskulären Natriumkanäle zugrunde, was zu wiederholten Muskelaktivierungen führt.
Die Myotonie bei myotonen Dystrophien ist Folge einer durch RNA-Toxizität gestörten Dysfunktion von Chlorid- und weiteren Ionenkanälen, wobei das gestörte Splicing des CLCN1 als Hauptmechanismus gilt.
Die Behandlung von Myotonien ist symptomatisch ausgerichtet, da eine Heilung derzeit nicht möglich ist.
Medikamentöse Therapie bei symptomatischer Myotonie: Bei leicht bis mäßig ausgeprägter Myotonie ist häufig keine medikamentöse Behandlung erforderlich, da Patienten lernen, mit der Erkrankung umzugehen. Bei ausgeprägter Myotonie oder beruflicher Behinderung sind Natriumkanal-Antagonisten die Medikamente der ersten Wahl.
Bei den multisystemischen Myotonien ist zu berücksichtigen, dass hier neben der Muskulatur auch andere Organe betroffen sein können, z.B. das Herz, Auge, Gehirn, Hormone, Stoffwechsel und innere Organe, so dass die medizinische Behandlung komplexer und interdisziplinär ist. Wichtig ist die angepasste Heil-und Hilfsmitteltherapie.